Silver-Russell syndrome: a case report
Bing-Ping Qiu, Chang-Hong Shi
Tengzhou, China
Author Affiliations: Department of Pediatrics, The Central Hospital of Tengzhou, Tengzhou 277500, China (Qiu BP, Shi CH)
Corresponding Author: Bing-Ping Qiu, MD, Department of Pediatrics, The Central Hospital of Tengzhou, Tengzhou 277500, China (Tel: 86-632-5626598; Email: qiuyangxiao@163.com)
Background: We present a case of an 8-year-old girl with a short stature. She had features of Silver-Russell syndrome (SRS) including asymmetrical body, maldevelopment of the fifth finger clinodactyly, small face, broad forehead, and delayed bone age. She had been diagnosed as having intrauterine growth retardation after birth, and feeding problems had been noted in her infancy. Psychomotor development was delayed.
Methods: The patient underwent physical examina-tion, X-ray examination, and laboratory examination. Treatment with recombinant human growth hormone (rhGH) and gonadotropin releasing hormone analogue (GnRHa) was given, and the patient was followed up.
Results: The imaging of the legs revealed that her left leg was 2 cm shorter and the circumference of the upper left leg was larger than the right one. During the follow-up for 2 years, her growth accelerated with mature breast and pubic hair. The levels of blood follicle stimulating hormone (FSH), luteinizing hormone (LH) and estradiol (E2) increased to the levels of adolescence.
Conclusions: This patient met the clinical criteria for SRS, and also with sexual precosity, she could be treated with rhGH and GnRHa, but the final height would not be ideal.
Key words: Silver-Russell syndrome; asymmetry; intrauterine growth retardation
World J Pediatr 2007;3(1):68-70
An 8-year-old girl was referred to the Central Hospital of Tengzhou because of short stature. She was the first term infant without birth asphyxia. At birth, her weight was 1500 g (<3rd percentile) and length was 41 cm (<3rd percentile). She had been diagnosed as having intrauterine growth retardation after birth. In the period of breast-feeding she cried easily, sweated, and suckled frequently. Psychomotor development was delayed. At 13 months she could sit without support and she started to walk at the age of 2.5 years. Her anterior fontanelle closed at 3 years old. Since walking on her own, she was found lame in her left leg.
Her mother had a history of catching cold at 2-month pregnancy, but no medication was prescribed. Her parents did not have a consanguineous marriage, and the family history was not positive for short stature.
In August 2003, the girl was brought to this hospital because of dwarf. Physical examination at that time revealed body height 101 cm, arm span 97 cm, sitting height 55.6 cm, head circumference 54.5 cm, asymmetrical body, broad forehead with a small triangular face, heavy eyebrows, and teeth badly arranged. Her thoracic cavity was normal, breast growth stage I (Tanner standard), auscultation of the heart and lung normal, and external genital organ normal. Her left leg was 2 cm shorter and the circumference of the upper left leg was larger than the right one. The activity of each joint was normal. Imaging of the legs (Fig. 1) showed that the left tibia was 19.8 cm long and the right tibia 21.8 cm, and their texture was normal. Imaging of the hands (Fig. 2) showed that the texture of the end of the first and fifth fingers was dysplastic, bending interiorly, and the bone age was 3.5 years. Head magnetic resonance imaging (MRI) and electroencephalography showed normal results. Laboratory examination showed normal functions of the thyroid, liver and kidney. Growth hormone stimulation test with insulin and levodopa showed that the levels of growth hormone were all <7 μg/L, blood follicle stimulating hormone (FSH) 6 U/L, luteinizing hormone (LH) 5.5 U/L. A diagnosis of Silver-Russell syndrome (SRS) was made clinically.[1]
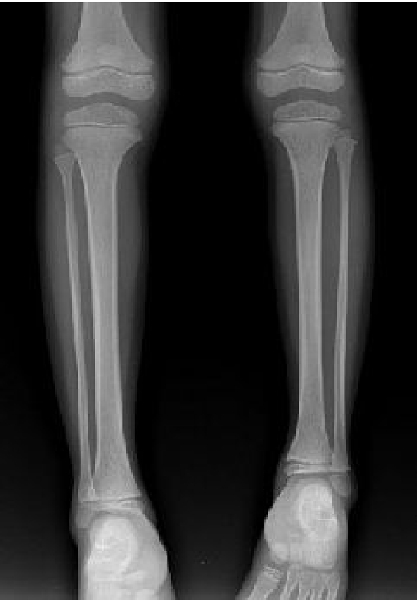
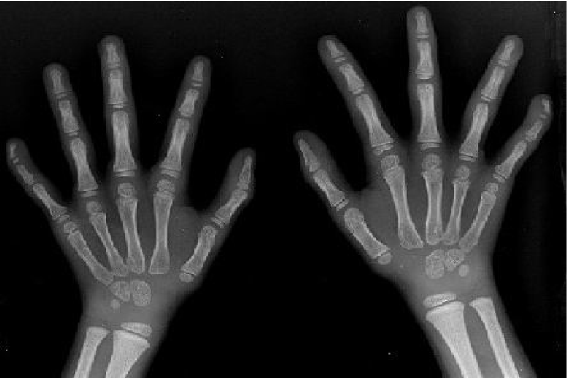
Fig. 1. The image of legs showing tibias not the same length, left tibia 19.8 cm, right tibia 21.8 cm, and the texture of the bones was normal.Fig. 2. The image of hands showing the texture of the end node of the fifth finger being dysplasia, bending toward interior.
In December 2004, her breast started to grow, and one year later she had no menstruation, pubic hair appeared, and growth accelerated. Physical examination showed body height 115 cm, arm span 112 cm, sitting height 62.5 cm, head circumference 55 cm, asymmetrical body, a small triangular face with a broad forehead, heavy eyebrows, small and narrow chin, and teeth badly arranged. Her thoracic cavity was normal, breast growth stage IIII (Tanner standard), auscultation of the heart and lung normal, normal external genital organ, and pubic hair growth stage II. X-ray revealed that her bone age was 6.5 years. Laboratory results included blood FSH 6.8 U/L, LH 8.7 U/L, and estradiol (E2) 65.88 pmol/L.
Discussion
Silver-Russell syndrome is also called asymmetry-dwarf-dysgenesis syndrome.[2] SRS was first described by Silver and colleagues in 1953 and later by Russell in 1954. The disease in children was characterized by facial abnormalities, low birth weight, asymmetrical body, and growth retardation.[3,4]
Clinically and genetically SRS is a heterogeneous disorder, and the underlying defect is unknown. Chromosome abnormalities have been found to be associated with the disease; among them chromosomes 7 and 17 are frequently involved. In 7% of sporadic cases, maternal uniparental disomy of chromosome 7 has been detected. Recent findings suggested that imprinting defects in the region of 11p15 also play a role in SRS.[5-8] Over the past several years, more than 400 patients with mild to classic phenotypes have been described. The estimated incidence of SRS ranged from 1 in 100 000 to 1 in 3000. Male and female children are equally affected.[9]
Pathophysiologically, growth failure is a primary abnormality. Patients typically present with intrauterine growth retardation, difficulty in feeding, failure to thrive, or postnatal growth retardation. Adequate catch-up growth often does not occur, and growth hormone insufficiency may be present. Abnormalities of spontaneous growth hormone secretion and subnormal responses to provocative growth hormone stimulation have been reported in a significant number of children with SRS.[10,11] Dysmorphism exists with small triangular face and normal head circumference. Because the length is usually less than normal, the head appears to be disproportionately large. With normal intelligence, the patient may have a learning disability somehow.[12-15]
The clinical characteristics of SRS include low birth weight, poor growth during infancy and childhood, short stature with normal skeleton and delayed bone age, normal head circumference, with the appearance of "pseudohydrocephalus", broad forehead with a small triangular face, small and narrow chin, limbs of different length, the curved fifth finger, and arm span being less than body height.[16] Genitourinary problems, gastrointestinal disorders, cardiac defects or malignancy may be present.[17-19]
No sexual precosity was seen during the patient's first visit to our hospital, but the levels of FSH and LH were higher than those of the average cohorts. This is probably related to her young age. After a follow-up for 2 years, her breast grew, growth accelerated, pubic hair appeared, blood FSH, LH and E2 increased to the levels of adolescents. These changes conformed to the diagnosis of sexual precosity, which confirmed the diagnosis of SRS.[1]
Growth hormone therapy is often considered for a child with SRS who has not manifested adequate catch-up growth at the age of 2 years. The use of growth hormone was approved by the US Food and Drug Administration in 2001 in children born small for gestational age who have not yet manifested adequate catch-up growth at the age of 2 years. Recombinant human growth hormone (rhGH) is given daily via subcutaneous injection at a dose of 0.48 mg/kg per week.[20,21] After treatment, body height may increase obviously in the first year, but decrease progressively year by year. Bone age tends to mature accelerately, and no conclusion has been reached on the influence of rhGH treatment on the final height of SRS patients.[22,23] Early intervention including physical therapy is beneficial, and special education courses are needed when the child is getting older.[24] The height of our patient was lower than that of children of the same age and sex, and she also had sexual precosity. Her final height would not be ideal, and treatment with rhGH and gonadotropin releasing hormone analogue, and follow-up were advisable.
Funding: None.
Ethical approval: Not needed.
Competing interest: None declared.
Contributors: QBP wrote the main body of the article. SCH provided advice on medical aspects. All authors contributed to interpretation of the study.
References
1 Price SM, Stanhope R, Garrett C, Preece MA, Trembath RC. The spectrum of Silver-Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J Med Genet 1999;36:837-842.
2 Li YX, Yan C. Endocrinology of pediatrics. Beijing: People's Health Publishing House, 1991: 49-50.
3 Silver HK, Kiyasu W, George J, Deamer WC. Syndrome of congenital hemihypertrophy, shortness of stature, and elevated urinary gonadotropins. Pediatrics 1953;12:368-375.
4 Russell A. A syndrome of "intra-uterine" dwarfism recognizable at birth with cranio-facial dysostosis, disproportionately short arms, and other anomalies (5 examples). Proc R Soc Med 1954;47:1040-1044.
5 Rossignol S. Silver-Russell syndrome and its genetic origins. J Endocrinol Invest 2006;29(1 Suppl):9-10.
6 van Haelst MM, Eussen HJ, Visscher F, de Ruijter JL, Drop SL, Lindhout D, et al. Silver-Russell phenotype in a patient with pure trisomy 1q32.1-q42.1: further delineation of the pure 1q trisomy syndrome. J Med Genet 2002;39:582-585.
7 Monk D, Bentley L, Hitchins M, Myler RA, Clayton-Smith J, Ismail S, et al. Chromosome 7p disruptions in Silver Russell syndrome: delineating an imprinted candidate gene region. Hum Genet 2002;111:376-387.
8 Langlois S, Yong SL, Wilson RD, Kwong LC, Kalousek DK. Prenatal and postnatal growth failure associated with maternal heterodisomy for chromosome 7. J Med Genet 1995;32:871-875.
9 Wollmann HA, Kirchner T, Enders H, Preece MA, Ranke MB. Growth and symptoms in Silver-Russell syndrome: review on the basis of 386 patients. Eur J Pediatr 1995;154:958-968.
10 Eggermann T, Wollmann HA, Kuner R, Eggermann K, Enders H, Kaiser P, et al. Molecular studies in 37 Silver-Russell syndrome patients: frequency and etiology of uniparental disomy. Hum Genet 1997;100:415-419.
11 Moore GE, Abu-Amero S, Wakeling E, Hitchins M, Monk D, Stanier P, et al. The search for the gene for Silver-Russell syndrome. Acta Paediatr Suppl 1999;88:42-48.
12 Jones KL. Russell-Silver syndrome. In: Smith's Recognizable Patterns of Human Malformation, 5th ed. Philadelphia: W. B. Sanders Company, 1997: 96-99.
13 Davies PS, Valley R, Preece MA. Adolescent growth and pubertal progression in the Silver-Russell syndrome. Arch Dis Child 1988;63:130-135.
14 Lai KY, Skuse D, Stanhope R, Hindmarsh P. Cognitive abilities associated with the Silver-Russell syndrome. Arch Dis Child 1994;71:490-496.
15 Schlegel D, Arcona A, Morgan J, Hatt C. Silver-Russell syndrome: a case study of neuropsychological deficits in an 8-year-old male. Arch Clin Neuropsychol 2000;15:790-791.
16 Eggermann T, Meyer E, Ranke MB, Holder M, Spranger S, Zerres K, et al. Diagnostic proceeding in Silver-Russell syndrome. Mol Diagn 2005;9:205-209.
17 Arai Y, Wakabayashi Y, Pak K, Tomoyoshi T. Horseshoe kidney in Russell-Silver syndrome. Urology 1988;31:321-323.
18 Ortiz C, Cleveland RH, Jaramillo D, Blickman JG, Crawford J. Urethral valves in Russell-Silver syndrome. J Pediatr 1991;119:776-778.
19 Anderson J, Viskochil D, O'Gorman M, Gonzales C. Gastrointestinal complications of Russell-Silver syndrome: a pilot study. Am J Med Genet 2002;113:15-19.
20 Stanhope R, Albanese A, Azcona C. Growth hormone treatment of Russell-Silver syndrome. Horm Res 1998;49 (Suppl 2):37-40.
21 Cutfield WS, Lindberg A, Rapaport R, Wajnrajch MP, Saenger P. Safety of growth hormone treatment in children born small for gestational age: the US trial and KIGS analysis. Horm Res 2006;65(Suppl 3):153-159.
22 Zeng JS, Wang DF. Endocrinology of modern pediatrics: foundation and clinics. Shanghai: Shanghai Science and Technology Publisher, 2000: 69.
23 Rizzo V, Traggiai C, Stanhope R. Growth hormone treatment does not alter lower limb asymmetry in children with Russell-Silver syndrome. Horm Res 2001;56:114-116.
24 Christoforidis A, Maniadaki I, Stanhope R. Managing children with Russell-Silver syndrome: more than just growth hormone treatment? J Pediatr Endocrinol Metab 2005;18:651-652.
Received September 14, 2006 Accepted after revision November 29, 200
