|
Intracranial solitary juvenile
xanthogranuloma in an infant
Lian-Ping Sun, Hui-Ming Jin, Bo Yang, Xiang-Ru Wu
Shanghai, China
Author Affiliations: Department of Pediatric Neurosurgery (Sun LP, Jin HM, Yang B); Department of Pathology (Wu XR), Xin Hua Hospital Affiliated to Shanghai Jiaotong University, Shanghai 200092, China
Corresponding Author: Lian-Ping Sun, MD, Department of Pediatric Neurosurgery, Xin Hua Hospital Affiliated to Shanghai Jiaotong University, Shanghai 200092, China (Tel: 86-21-65790000 ext 8285; Email: slp1128@126.com)
Background: Juvenile xanthogranuloma (JXG) is a disorder of histiocyte proliferation. Most cases present with a solitary cutaneous lesion. JXG with systemic involvement is rare with significant morbidity. Intracranial solitary JXG may be misdiagnosed before operation.
Methods: A 5-month-old boy showed an elevated anterior fontanel but no other abnormalities on admission. Brain MRI showed a large mass in the right parietal region.
Results: The tumor was removed with the encroached meninges. A JXG in the right parietal region was diagnosed pathologically.
Conclusion: Total excision of the tumor may be curative with a prerequisite of ensuring normal vital signs and nervous function.
Key words: infant; intracranial; juvenile xanthogranuloma; surgery
World J Pediatr 2009;5(1):71-73
Introduction
Juvenile xanthogranuloma (JXG) is a disorder of histiocyte proliferation. Many patients present with cutaneous maculopapular lesions. JXG with systemic (extracutaneous) involvement is rare with significant morbidity. The most frequent extracutaneous sites involved are the central nervous system, liver, spleen, lungs, kidneys, eyes, etc. To date, few JXG cases were reported with single organ involvement but without cutaneous lesions, and even fewer cases were demonstrated as intracranial solitary JXG, which may be misdiagnosed before operation. Herein we report a case of intracranial solitary JXG in an infant.
Case report
A 5-month-old boy was admitted to the hospital for his elevated anterior fontanel which was revealed before he was referred to our department. On admission, he was conscious and neurological examination was normal. Physical examination showed an abnormal anterior fontanel about 2.5×2.5 cm2 and his head circumference was 42 cm. There were no abnormalities in the skin, liver, spleen, and lungs. The result of blood examination was normal.
MRI demonstrated a large heterogeneous high-intensity lesion with smooth margins located in the right parietal region (Fig. 1). The lesion was about 4.5×4.5×3.5 cm3 in size with the contiguous enhanced dura and an equal signal on T1- and T2-weighted images respectively. Contrast enhancement scanning showed that the lesion had a notably enhanced signal without visible edema around it and the basilar part of it was adhered to the meninges. According to the radiologic findings, a brain tumor in the right parietal region was diagnosed preoperatively. The patient underwent a neurosurgical procedure in our department.
As demonstrated in surgery, the inner plate of the parietal bone was oppressed by the tumor and became partially moth-eaten. The size of the encroached meninges and the tumor was about 3×3 cm2 and 5×5×4 cm3 respectively. The tumor was round, flesh pink color and extended into the brain and was separated clearly from the brain tissue by its peplos. There was abundant blood supply to the tumor, which mainly came from dural arteries. Then, these vessels were blocked by electric coagulation and the tumor was totally removed with the encroached meninges.
Microscopic examination revealed that the tumor was composed of Touton giant cells (Fig. 2A), large amounts of foam cells, eosinophile granulocytes and small amounts of macrophages. Postoperative immunohistochemistry showed that the cells were CD68+ (Fig. 2B), Cdla(-) (Fig. 2C) and S100(-). A JXG in the right parietal region was diagnosed. MRI showed no recurrence of xanthogranuloma 6 months after surgery (Fig. 3).
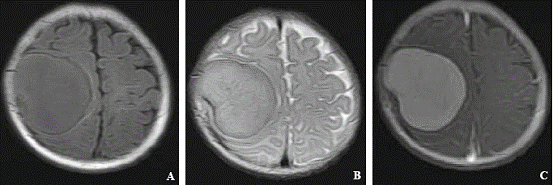
Fig. 1. MRI demonstrating a mass in the right parietal region. A: T1-weighted; B: T2-weighted; C: contrast-enhanced.
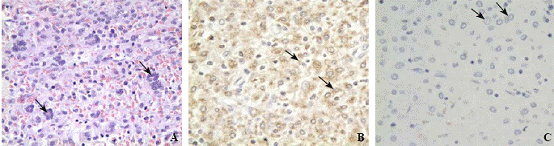
Fig. 2. A: The tumor consisting Touton giant cells, foam cells, etc (original magnification × 400); B: Immunohistochemistry indicating CD68(+) (original magnification × 400); C: Immunohistochemistry showing Cdla(-) (original magnification × 400).
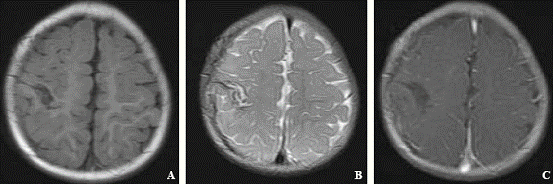
Fig. 3. Follow-up MRI demonstrating no xanthogranuloma recurrence 6 months after surgery. A: T1-weighted; B: T2-weighted; C: contrast-enhanced.
Discussion
JXG is clinically classified into the cutaneous form and the systemic form. Cases of systemic JXG have been reported.[1,2] Systemic JXG causes cutaneous lesions with extracutaneous involvements frequently seen in the central nervous system, liver, spleen, lung, kidneys and bones. It is rare to see a brain tumor with no cutaneous lesion, and there are 16 cases reported so far.[3-8]
JXG is a specialized proliferation of non-Langerhans' cells.[9] It is not a real tumor but a course of reactive proliferation of histiocytes caused by some stimulants such as viral infection and physical factors and often be confused with Langerhan's cells hyperplasia (LCH) clinically. The identification of JXG from LCH mainly depends on pathological examinations.[10] The typical histiocytic compositions of JXG include Touton giant cells, foam cells, and foreign body giant cells, except lymphocytes and eosinophile granulocytes. Ultrastructurally, JXG presents no Berbeck particles. Immunohistochemistry examination usually demonstrates the following: CD1a(-), S100(-), CD68(+), Vimentin(+), VIIIa(+), HAM56(+) and LYS(+). Approximately 2/3 patients with JXG are the infants who are younger than 6 months after birth and nearly 10% are adults. JXG often involves skin tissues and mucosa, which appears yellow, jacinth mottling or as larger parenchymatous nodus, or papula, or simultaneously involves internal organs. Complete surgical excision is feasible for therapy of symptomatic lesions, whereas external beam irradiation and multiagent chemotherapy are utilized for unresectable masses. Cutaneous lesions have the possibility of self-limited process. If important organs are not damaged for months or years, JXG may spontaneously regressed.
The central nervous system involvement might be considered a separate entity due to its mortality.[11] Its pathological changes in the brain may be originated from mesenchymal stem cells of the dura mater and from the intracerebral perivascular soft tunica vaginalis or the wall of brain vessels themselves. The lesion can be a simple or multiple involvement. In the brain tissue, meninges, choroid plexus or cranial nerve, JXG can induce epilepsy, mental and growth retardation, diabetes insipidus, ataxia, hemiplegia. According to the literatures, JXG appears in the central nervous system secondary to the skin, soft tissues, lung, pancreas which have been involved for several months or years. It is rare for the cases of intracranial JXG only without cutaneous manifestations as the present case. It is difficult to differentiate the tumor from meningioma and glioma by preoperative radiological findings, but postoperative pathological examination demonstrated that there were a lot of foam cells as well as Touton giant cells in the tumor. The immunohistochemistry findings of S100(-), CD68(+) and CD1a(-) also support the diagnosis of JXG. With regard to a single tumor, it is necessary to resect the tumor totally to save the patient or to release the compression to nerves. If the tumor locates at the surgically inaccessible location or there are multicentric lesions, the patient will still have a eusemia with stereotactic radiosurgery[10] or the therapeutic alliance[12] in time with cytarabine, vincristine, MTX, prednisolone (or hydrocortisone).
Funding: None.
Ethical approval: Not needed.
Competing interest: None declared.
Contributors: Sun LP wrote the main body of the article. All authors contributed to the design and interpretation of the study.
References
1 Freyer DR, Kennedy R, Bostrom BC, Kohut G, Dehner LP. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr 1996;129:227-237.
2 Tan MH. Juvenile xanthogranuloma of the pancreas: a case report and review of literature. J Chin Clin Med Abstract 2004;1:25-26.
3 Gutnik H, Dolenc-Strazar Z, Popovic M. September 2000: 15 year old girl with intracranial lesion. Brain Pathol 2001;11: 123-125.
4 Schultz KD Jr, Petronio J, Narad C, Hunter SB. Solitary intracerebral juvenile xanthogranuloma. Case report and review of the literature. Pediatr Neurosurg 1997;26:315-321.
5 Ashley WW Jr, Narayan P, Park TS, Tu PH, Perry A, Leonard JR. Incidental pediatric intraparenchymal xanthogranuloma: case report and review of the literature. J Neurosurg 2005; 102(3 Suppl):307-310.
6 Jung CS, Schänzer A, Hattingen E, Plate KH, Seifert V. Xanthogranuloma of the sellar region. Acta Neurochir (Wien) 2006;148:473-477.
7 Pimentel J, Fernandes A, Távora L, Miguéns J, Lobo Antunes J. Benign isolated fibrohistiocytic tumor arising from the central nervous system. Considerations about two cases. Considerations about two cases. Clin Neuropathol 2002;21:93-98.
8 Nakasu S, Tsuji A, Fuse I, Hirai H. Intracranial solitary juvenile xanthogranuloma successfully treated with stereotactic radiosurgery. J Neurooncol 2007;84:99-102.
9 Ladisch S. Histiocytosis syndrome of childhood. Nelson textbook of pediatrics, 17th ed. An Imprint of Elsevier Science, 1727-1730.
10 Eggli KD, Caro P, Quiogue T, Boal DK. Juvenile xantho-granuloma: non-X histiocytosis with systemic involvement. Pediatr Radiol 1992;22:374-376.
11 Ernemann U, Skalej M, Hermisson M, Platten M, Jaffe R, Voigt K. Primary cerebral non-Langerhans cell histiocytosis: MRI and differential diagnosis. Neuroradiology 2002;44: 759-763.
12 Nakatani T, Morimoto A, Kato R, Tokuda S, Sugimoto T, Tokiwa K, et al. Successful treatment of congenital systemic juvenile xanthogranuloma with Langerhans cell histiocytosis-based chemotherapy. J Pediatr Hematol Oncol 2004;26:371-374.
Received October 12, 2007 Accepted after revision March 26, 2008
|
