|
Congenital pulmonary lymphangiectasis
Zuo-Yuan Xiao, Yu Tao, Xin-Yi Tang, Guo-Juan Chen, Lei Guo
Guangzhou, China
Author Affiliations: Department of General Pediatrics, The Third Affiliated Hospital of Sun Yat-sen University (Xiao ZY, Tang XY, Chen GJ, Guo L); Department of Pathology, The First Affiliated Hospital of Sun Yat-sen University (Tao Y), Guangzhou, China
Corresponding Author: Zuo-Yuan Xiao, Department of General Pediatrics, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou 510630, China (Tel: 86-20-85253098; Email: xiaozy2@126.com)
Background: Congenital pulmonary lymphangiectasis (CPL) characterized by dilatation of pulmonary lymphatic vessels occurs as a congenital anomaly. With poor prognosis, neonatal presentation of bilateral CPL is associated with the severe compromise of pulmonary gas exchange and high mortality.
Methods: A male infant born at 39 weeks of gestation was found to have CPL. Cyanosis and cardiac arrest occurred a few minutes after birth, and the symptoms remained after artificial ventilation. The infant died of hypoxemic cardiac failure 45 minutes after birth. Autopsy showed neither pleural effusion nor valvular abnormalities.
Results: Microscopically dilated vessels with lymphatics were seen in the lung of the infant. Atelectasis, CPL, inhalation of amniotic fluid, partial hydropic degeneration of hepatic cells, and scrotal edema were diagnosed.
Conclusion: With regard to treatment and prognosis, CPL must be distinguished from interstitial emphysema and other diseases.
Key words: congenital disease; neonate; pulmonary lymphangiectasis
World J Pediatr 2009;5(1):68-70
Introduction
Congenital pulmonary lymphangiectasis (CPL) is characterized by the increasing number of lymphatic vessels and the cystic dilatation of pulmonary lymphatic vessels in the interstitium.[1] With poor prognosis, neonatal presentation of bilateral CPL is associated with the severe compromise of pulmonary gas exchange and high mortality.[2] Because of its aggressive course, the disease tends to be misdiagnosed as interstitial emphysema. With regard to treatment and prognosis, CPL must be distinguished from interstitial emphysema and other diseases.[3]
Case report
A male infant born at 39 weeks of gestation after a cesarean section was the first of the non-consanguineous parents. Prenatal diagnosis including ultrasonography showed normal results. The amniotic fluid was clear with an amount of 400 ml. The infant weighed 3.0 kg with apgar scores of 6 and 7 at 1 and 5 minutes, respectively. The infant was pink at birth, with normal muscular tension and no edema, while he failed to establish spontaneous breath with cyanosis, then hypermyotonia and cardiac arrest were noticed. Immediate treatment included stimulating the infant to breathe by cardiac compression and artificial ventilation. Then the infant was intubated, and intravenous administration with nitroglycerin was given. The symptom of cyanosis did not improve. And some white chyliform liquid gushed out from the nasal cavity after the intubation. Gasbag was used to pump oxygen into the lung, but strong resistance was noted. The infant died of hypoxemic cardiac failure 45 minutes after birth.
Post-mortem examination of the thoracic cavity revealed normal size of the thymus and no fluid in the pleural cavity. The left lung occupied approximately 1/3 of the thoracic cavity, which was divided into two lobes sized 6.0×3.5×2.5 cm3 (the normal average size of the same weight is 6.6×5.5×1.6 cm3)[4]. The density of the lung was higher than fresh water. The right lung occupied approximately 1/2 of the thoracic cavity and divided into three lobes sized 8.0×3.5×2.5 cm3 (the normal average size of the same weight was 7.4×6.0×1.9 cm3)[4]. The density of the lung was lower than fresh water.
Below the lung membrane, cystic-like tissues could be seen ranging from 0.1×0.1 cm2 to 0.3×0.5 cm2, especially in the right lung, but the primary bronchus was normal. Neither pericardial effusion nor pericardiosymphysis could be found. The heart showed normal structure and size without consolidations. No abnormalities of valves, atrial septum or interventricular septum were identified. The throat and esophagus had no abnormal pathological change.
Figs. 1-4 show the histological changes of the lung and bronchi. Interstitium congestion was found in the liver, spleen, kidney and adrenal gland, and hydropic degeneration in some hepatic cells.
Primary pathological changes included inhalation of amniotic fluid, interstitial emphysema, atelectasis, and scrotal edema. The final diagnosis was made of atelectasis, CPL, inhalation of amniotic fluid, hydropic degeneration of some hepatic cells, and scrotal edema.
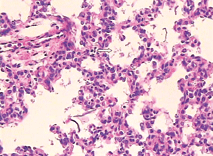 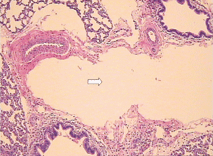
Fig. 1. Part of pulmonary alveolus collapsed and alveolar wall close to each other (HE staining, original magnification × 40). Fig. 2. Some amniotic fluid-like material inside the pulmonary alveolus (HE staining, original magnification × 400).
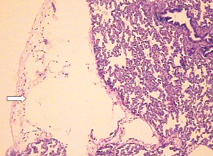 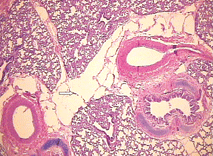
Fig. 3. Microscopic features of the lung showing cystic dilatation of the lymphatics (HE staining, original magnification × 100). Fig. 4. Cystic dilatation of the lymphatics (HE staining, original magnification × 100).
Discussion
CPL characterized by dilatation of the pulmonary lymphatic vessels occurs as a congenital anomaly. It is sporadic but some cases of familial CPL have been recognized.[5] Half of the cases are still born, and the rest die of respiratory distress at the birth or within the few hours after birth. The first case of CPL reported by Virchow in 1856 showed such manifestations as aggressive dyspnea after birth, recession and cyanosis.[1] X-ray examination showed atelectasis, punctiform or reticulate shadows, frosted-glass like infiltration in both lungs. Hence it is occasionally difficult to differentiate CPL from interstitial pneumonitis and hyaline membrane disease. Because of the increased number of lymph tubes and dilation of lymphatic tubes, CPL is difficult to be differentiated from interstitial emphysema. A definitive diagnosis of CPL can only be made by autopsy.
The first case of CPL in China reported by Zhu et al[6] from Nanjing General Hostpital of Nanjing Military Command was misdiagnosed as interstitial pulmonary emphysema at autopsy. The correct diagnosis of CPL could only be made until the pulmonary phlebemphraxis was found at biopsy.[6,7] Pulmonary lymphangiectasis is divided into primary (congenital) and secondary types. The primary type presents in neonates and is usually fatal. The secondary results from a variety of processes that impair lymph drainage and increase lymph production. Primary lymphangiectasis probably results from a failure of pulmonary interstitial connective tissues, leading to dilation of the pulmonary lymphatic capillaries. Secondary lymphangiectasis may be due to surgery, radiation, infection, tumor genesis and development of congenital heart diseases. Nowadays both of them are usually named as congenital pulmonary lymphangiectasis. Noonan et al[8] classified CPL into three types: type 1 is a generalized form of lymphangiectasis; type 2 is caused by pulmonary venous hypertension or obstruction associated with cardiovascular anomalies; and type 3 is due to a primary developmental defect of the lung lymphatics. Type 1 and 3 CPL are congenital and type 2 CPL is secondary. Type 3 is associated with an aggressive course and poor prognosis.[8,9] Patients with type 1 CPL have better prognosis. Most of type 2 patients may die in fetal stage or only a few days after birth. Most of the type 3 patients who have a worse prognosis are male, and likely die of respiratory distress. Moerman et al[10] reported 7 cases of primary or congenital lymphangiectasis complicated with bilateral chylothorax at perinatal autopsy. They found that the causes of CPL are varied and the primary pathological change is the obstruction of lymphatic ducts caused by the maldevelopment of the lymphatic system but not by the anomaly of the lung. Regardless of the causes of CPL, the dilation of pulmonary lymphatic ducts induces sclerosis of the lung and reduces the compliance of the lung, leading to respiratory distress. In the present case, there were no lymphatic dilatations in the heart, liver and spleen. It was categorized as type 1 CPL because of the aggressive course and severe lymphatic dilatations of the lung. In a neonate with CPL reported by Bachiri et al,[11] cardiac arrest was the only symptom, which was similar to that found in the present case. In this case, pulmonary alveolus could not distend adequately, and oxygen could not be pumped into the lung under a large force of resistance. Thus we consider that lymphangiectasis and lung anomaly may cause the sclerosis of the lung. Compared with pulmonary lymphangiectasis, interstitial emphysema is the secondary pathological change caused by artificial ventilation, much air forced to the pulmonary alveoli, resulting in their collapse and then the penetration of air into the loose connective tissue near the lobulus pleura and bronchus.[12] There is no endotheliocyte in the dilated cyst, which could help to distinguish it from CPL.
With regard to treatment and prognosis, CPL should be distinguished from interstitial pneumonitis, hyaline membrane disease, and interstitial emphysema.
Funding: None.
Ethical approval: Not needed.
Competing interest: None declared.
Contributors: Xiao ZY wrote the first draft of the paper. All authors contributed to the intellectual content and approved the final version. Tang XY is the guarantor.
References
1 Laurence KM. Congenital pulmonary cystic lymphangiectasis. J Pathol Bacteriol 1955;70:325-333.
2 Hoehn T, William M, McPhaden AR. Endothelial, inducible and neuronal nitric oxide synthase in congenital pulmonary lymphangiectasis. Eur Respir J 2006;27:1311-1315.
3 Finder J, Steinfeld J. Congenital pulmonary lymphangiectasia. N Engl J Med 2004;350:948.
4 Shi SZ, Huang PW, Wang SZ, Zhu MJ, Gu XY, Gao ES. The weights and size of various organs in 940 newborns. Shanghai Med Journal 1981;4:31-36.
5 Hirano H, Nishigami T, Okimura A, Nakasho K, Kunio Uematsu. Autopsy case of congenital pulmonary lymphangiectasis. Pathol Int 2004;54:532-536.
6 Zhu QY, Jin CH, Li FS, Wu WP. Cogenital pulmonary lymphangiectasis. Chin J Pathol 1995;24:80-82.
7 Laurence KM. Congenital pulmonary lymphangiectasis. J Clin Pathol 1959;12:62-69.
8 Noonan JA, Walters LR, Reeves JT. Congenital pulmonary lymphangiectasis. Am J Dis Child 1970;120:314-319.
9 Gilewski MK, Statler CC, Kohut G, Toriello HV. Congenital pulmonary lymphangiectasia and other anomalies in a child: provisionally unique syndrome? Am J Med Genet 1996;66:438-440.
10 Moerman P, Vandenberghe K, Devlieger H, Van Hole C, Fryns JP, Lauweryns JM. Congenital pulmonary lymphangiectasis with chylothorax; a heterogeneous lymphatic vessel abnormality. Am J Med Gernrt 1993;47:54-58.
11 Bachiri A, Djebara A, Klosowski S, Haouari N, Thelliez P, Voisin O, et al. Congenital pulmonary lymphangiectasia revealed by cardiac arrest. Arch Pediatr 2003;10:615-618.
12 Xu YH, Zhou R. Newborn emphysema attributed to medical actions (3 case report). Fa Yi Xue Za Zhi 2006;22:133-134.
Received July 23, 2007 Accepted after revision April 10, 2008
|
